









Innehåll
Talassemi
Thalassemia är en uppsättning ärftliga blodsjukdomar som påverkar produktionen av hemoglobin (proteinet som ansvarar för transport av syre). De varierar i svårighetsgrad: vissa orsakar inga symtom medan andra är livshotande. En benmärgstransplantation övervägs i de allvarligaste fallen.
Thalassemi, vad är det?
Definition av talassemi
Thalassemi kännetecknas av en defekt i produktionen av hemoglobin. Som en påminnelse är hemoglobin ett stort protein som finns i röda blodkroppar (röda blodkroppar) vars roll är att säkerställa transporten av dixoygen från andningsorganen till resten av kroppen.
Det sägs att talassemi är en sjukdom i blodet. Röda blodkroppars transportfunktion är försämrad, vilket kan få allvarliga konsekvenser för kroppen. Vid denna tidpunkt är det viktigt att notera att det finns flera typer av talassemi som inte har samma egenskaper eller samma svårighetsgrad. Vissa har inga symtom medan andra är livshotande.
Orsaker till talassemi
Thalassemia är genetiska sjukdomar. De beror på förändringen av en eller flera gener involverade i syntesen av hemoglobin, och mer exakt på förändringen av generna som är involverade i produktionen av hemoglobinproteinkedjor. Det finns fyra av dessa: två alfa-kedjor och två beta-kedjor.
Var och en av dessa kedjor kan påverkas vid talassemi. Vi kan också urskilja:
- alfa-talassemi kännetecknad av en förändring av alfakedjan;
- beta-talassemi som kännetecknas av en förändring av betakedjan.
Svårighetsgraden av alfa-talassemi och beta-thalassemi beror på antalet förändrade gener. Ju viktigare det är, desto större svårighetsgrad.
Diagnos av talassemi
Diagnosen talassemi ställs genom blodprov. Det fullständiga blodvärdet gör det möjligt att utvärdera utseendet och antalet röda blodkroppar och därmed veta den totala mängden hemoglobin. Biokemiska analyser av hemoglobin gör det möjligt att skilja alfa-talassemi från beta-talassemi. Slutligen gör genetiska analyser det möjligt att utvärdera antalet förändrade gener och därmed definiera svårighetsgraden av talassemi.
Berörda personer
Thalassemia är ärftliga genetiska sjukdomar, det vill säga överförs från föräldrar till deras barn. De når främst människor från Medelhavskanten, Mellanöstern, Asien och Afrika söder om Sahara.
I Frankrike uppskattas förekomsten av alfa-talassemi till 1 av 350 personer. Incidensen av beta-talassemi uppskattas till 000 födslar per 1 och år över hela världen.
Symtom på talassemi
Symtomen på talassemi varierar mycket från fall till fall, och beror främst på graden av förändring av generna som är involverade i produktionen av hemoglobinproteinkedjor. Thalassemia kan vara symtomfria i sina mindre former och vara livshotande i sina mer allvarliga former.
Symtomen som nämns nedan gäller endast de mellanliggande till huvudformerna av talassemi. Dessa är bara de viktigaste symptomen. Mycket specifika symtom kan ibland ses beroende på typen av talassemi.
Anemi
Det typiska tecknet på talassemi är anemi. Detta är en brist på hemoglobin som kan resultera i uppkomsten av olika symtom:
- trötthet
- andnöd;
- blekhet;
- obehag;
- hjärtklappning.
Intensiteten av dessa symtom varierar beroende på svårighetsgraden av talassemin.
Gulsot
Personer med talassemi kan ha gulsot (gulsot) som är synligt på huden eller ögonvitan.
gallsten
Stenbildning inuti gallblåsan kan också ses. Beräkningar är som "små stenar".
splenomegali
Splenomegali är en förstoring av mjälten. En av rollerna för detta organ är att filtrera blodet och filtrera skadliga ämnen inklusive onormala röda blodkroppar. Vid talassemi är mjälten starkt mobiliserad och ökar gradvis i storlek. Smärta kan kännas.
Andra, mer sällsynta symtom
Mer sällan kan allvarliga former av talassemi leda till andra avvikelser. Till exempel kan det observeras:
- hepatomegali, det vill säga en ökning av leverns storlek;
- bendeformiteter;
- försenad barnutveckling;
- sår.
Hanteringen av talassemi är avgörande för att begränsa förekomsten av dessa komplikationer.
Behandlingar för talassemi
Hanteringen av talassemi beror på många parametrar inklusive typen av talassemi, dess svårighetsgrad och tillståndet hos den berörda personen. De mest mindre formerna kräver ingen behandling medan de svåra formerna kräver mycket regelbunden medicinsk övervakning.
De behandlingar som nämns nedan gäller endast de mellanliggande till större formerna av talassemi
Korrigering av anemi
När bristen på hemoglobin är för stor är regelbundna blodtransfusioner nödvändiga. De går ut på att injicera den berörda personen med blod eller röda blodkroppar som tagits från en givare för att upprätthålla en acceptabel nivå av röda blodkroppar i blodet.
Vitamin B9 tillskott
Det kan rekommenderas att börja dagliga vitamin B9-tillskott eftersom behovet av detta vitamin ökar vid talassemi. Vitamin B9 är involverat i produktionen av röda blodkroppar.
splenektomi
En splenektomi är kirurgiskt avlägsnande av mjälten. Denna operation kan övervägas när anemin är mycket viktig.
Behandling av järnöverskott
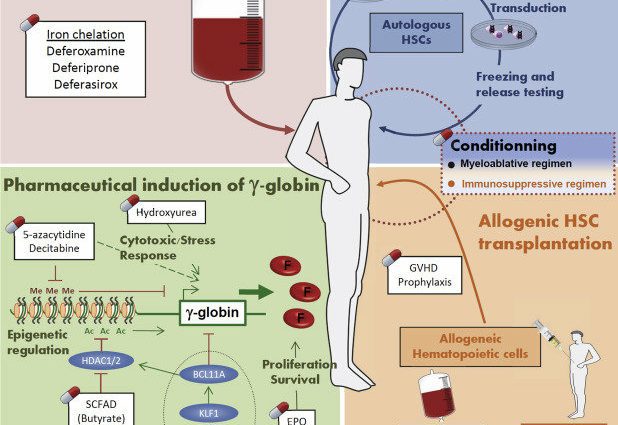
Personer med talassemi har en överbelastning av järn i kroppen. Denna ansamling kan leda till olika komplikationer. Det är därför järnkelatorer erbjuds för att ta bort överflödigt järn.
Benmärgstransplantation
En benmärgstransplantation är den enda behandlingen som permanent kan bota talassemi. Detta är en tung behandling som endast erbjuds vid de allvarligaste formerna av sjukdomen.
Förhindra talassemi
Thalassemi är en ärftlig genetisk sjukdom. Det finns ingen förebyggande åtgärd.
Däremot gör genetiska tester det möjligt att upptäcka friska bärare (personer som har en eller flera förändrade gen(er) men som inte är sjuka). Ett par friska bärare bör informeras om risken att föda ett barn med talassemi. I vissa fall kan denna risk bedömas av en genetiker. Prenatal diagnos kan också övervägas under vissa förutsättningar. Det bör diskuteras med din läkare.