









Innehåll
Bourneville tuberös skleros
Vad är det ?
Bourneville tuberös skleros är en komplex genetisk sjukdom som kännetecknas av utvecklingen av en godartad (icke-cancerös) tumör i olika delar av kroppen. Dessa tumörer kan sedan lokaliseras i huden, hjärnan, njurarna och andra organ och vävnader. Denna patologi kan också orsaka allvarliga problem i utvecklingen av individen. Men de kliniska manifestationerna och svårighetsgraden av sjukdomen varierar från patient till patient.
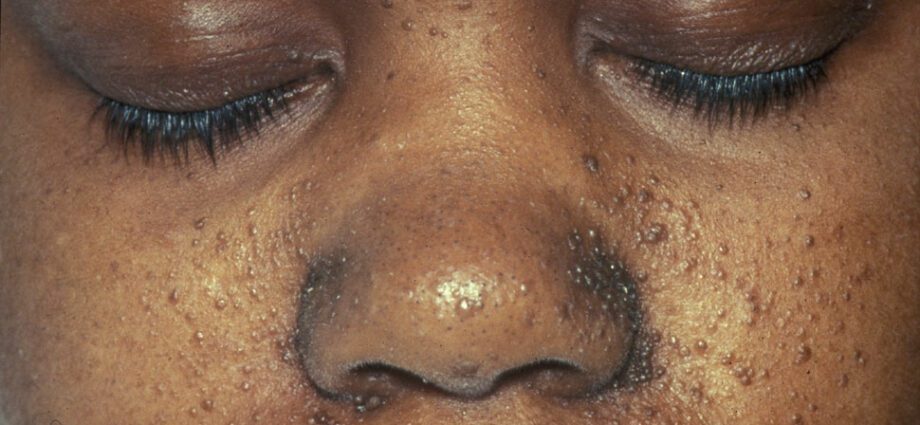
De associerade hudavvikelserna liknar i allmänhet fläckar på huden eller områden där huden är ljusare än på resten av kroppen. Utvecklingen av tumörer i ansiktet kallas angiofibrom.
I samband med hjärnskador är de kliniska tecknen epileptiska anfall, beteendeproblem (hyperaktivitet, aggressivitet, intellektuella funktionsnedsättningar, inlärningsproblem, etc.). Vissa barn med sjukdomen har till och med någon form av autism, utvecklingsstörningar, som påverkar sociala interaktioner och kommunikation. Godartade hjärntumörer kan också orsaka komplikationer som kan vara dödliga för patienten.
Utvecklingen av tumörer i njurarna är vanligt hos personer med tuberös skleros. Detta kan orsaka allvarliga komplikationer i njurfunktionen. Dessutom kan tumörer utvecklas i hjärtat, lungorna och näthinnan. (2)
Det är en sällsynt sjukdom, vars prevalens (antal fall i en given population vid en given tidpunkt) uppgår till 1/8 till 000/1 personer. (15)
Symptom
De kliniska manifestationerna i samband med tuberös skleros i Bourneville varierar beroende på de organ som påverkas. Dessutom varierar symtomen förknippade med sjukdomen mycket från en individ till en annan. Med symtom som sträcker sig från lindriga till svåra.
De mest kända symptomen på denna sjukdom inkluderar epileptiska anfall, kognitiva och beteendestörningar, hudavvikelser etc. De organ som oftast drabbas är: hjärnan, hjärtat, njurarna, lungorna och huden.
Utvecklingen av maligna (cancerösa) tumörer är möjlig vid denna sjukdom men är sällsynta och påverkar främst njurarna.
De kliniska tecknen på sjukdomen i hjärnan kommer från attacker på olika nivåer:
– skador på kortikala tuberkler;
– ependymala knölar (SEN);
– gigantiska ependymala astrocytom.
De resulterar i: utveckling av mental retardation, inlärningssvårigheter, beteendestörningar, aggressivitet, uppmärksamhetsstörningar, hyperaktivitet, tvångssyndrom, etc.
Njurskador kännetecknas av utvecklingen av cystor eller angiomyolipom. Dessa kan leda till njursmärta och till och med njursvikt. Om kraftig blödning märks kan det bero på svår anemi eller högt blodtryck. Andra allvarligare men sällsynta konsekvenser kan också vara synliga, särskilt utvecklingen av karcinom (tumör i de ingående cellerna i epitelet).
Ögonskador kan likna synliga fläckar på näthinnan och orsaka synstörningar eller till och med blindhet.
Hudavvikelser är många:
– hypomelaniska fläckar: som resulterar i uppkomsten av ljusa fläckar på huden, var som helst på kroppen, till följd av en brist på melanin, ett protein som ger huden färg;
- uppkomsten av röda fläckar i ansiktet;
– missfärgade fläckar på pannan;
– andra hudavvikelser, beroende från en individ till en annan.
Lungskador finns hos 1/3 av patienterna med en lätt kvinnlig övervikt. De associerade symtomen är då mer eller mindre allvarliga andningssvårigheter.
Ursprunget till sjukdomen
Sjukdomens ursprung är genetiskt och ärftligt.
Överföring involverar mutationer i generna TSC1 och TSC2. Dessa gener av intresse kommer in i bildandet av proteiner: hamartin och tuberin. Dessa två proteiner gör det möjligt att genom ett interaktivt spel reglera cellproliferation.
Patienter med sjukdomen föds med minst en muterad kopia av dessa gener i var och en av sina celler. Dessa mutationer begränsar sedan bildningen av hamartin eller tubertin.
I det sammanhang där de två kopiorna av genen är muterade förhindrar de helt produktionen av dessa två proteiner. Denna proteinbrist tillåter därför inte längre kroppen att reglera tillväxten av vissa celler och leder i denna mening till utvecklingen av tumörceller i olika vävnader och/eller organ.
Riskfaktorer
Riskfaktorerna för att utveckla en sådan patologi är genetiska.
Faktum är att överföringen av sjukdomen är effektiv genom ett autosomalt dominant läge. Antingen är den muterade genen av intresse lokaliserad vid en icke-sexuell kromosom. Dessutom är närvaron av endast en av de två kopiorna av den muterade genen tillräcklig för att sjukdomen ska utvecklas.
I denna mening har en individ som har en av dessa två föräldrar som lider av sjukdomen 50 % risk att själv utveckla den sjuka fenotypen.
Förebyggande och behandling
Diagnosen av sjukdomen är först och främst differentiell. Den bygger på atypiska fysiska kriterier. I de flesta fall är de första karaktäristiska tecknen på sjukdomen: förekomsten av återkommande epileptiska anfall och förseningar i patientens utveckling. I andra fall resulterar dessa första tecken i hudfläckar eller identifiering av en hjärttumör.
Efter denna första diagnos är ytterligare undersökningar nödvändiga för att validera diagnosen eller inte. Dessa inkluderar:
– en hjärnskanning;
– en MRT (magnetisk resonanstomografi) av hjärnan;
– ett ultraljud av hjärta, lever och njurar.
Diagnosen kan vara effektiv vid barnets födelse. I övrigt är det viktigt att det genomförs så snabbt som möjligt för att ta hand om patienten så snart som möjligt.
För närvarande finns det inget botemedel mot sjukdomen. De associerade behandlingarna är därför oberoende av de symptom som varje individ presenterar.
Vanligtvis ges antiepileptika för att begränsa anfall. Dessutom ordineras också läkemedel för behandling av tumörceller i hjärnan och njurarna. I samband med beteendeproblem är specifik behandling av barnet nödvändig.
Behandlingen av sjukdomen är vanligtvis långvarig. (1)